Οι αυτοάνοσες παθήσεις προκαλούνται από βλάβη οργάνων ή ιστών ή κυττάρων λόγω δυσλειτουργίας του ανοσοποιητικού μας συστήματος, που δυστυχώς εκλαμβάνει σαν ξένες ορισμένες πρωτεΐνες του σώματος μας και επιτίθεται εναντίον τους.
>> Οι αυτοάνοσες παθήσεις οφείλονται σε γονιδιακές μεταλλάξεις ή/και σε επιγενετικές (περιβαλλοντικές) αιτίες. (Δες στο τέλος)
Οι επιγενετικές αιτίες μπορεί να είναι μια λοίμωξη από ιό ή μικρόβιο, ορισμένα χημικά στη διατροφή ή στον εισπνεόμενο αέρα, ορισμένα φάρμακα, αλλαγές στο μικροβίωμα του εντέρου (λόγω της σύγχρονης υγιεινής διαβίωσης), ένα οξειδωτικό stress κλπ.
Το τελικό αποτέλεσμα είναι ότι ορισμένα από τα Τ λεμφοκύτταρα μας ή/και ορισμένα από τα Β λεμφοκύτταρα μας στρέφονται εναντίον του οργανισμού μας. Τα Β λεμφοκύτταρα προκαλούν αυτοάνοσες παθήσεις μέσω των αυτο-αντισωμάτων που παράγουν (αφού μετατραπούν σε πλασματοκύτταρα). (Δες στο τέλος)
ΛΙΓΑ ΓΙΑ ΤΗΝ ΕΞΟΥΔΕΤΕΡΩΣΗ ΟΣΩΝ ΛΕΜΦΟΚΥΤΤΑΡΩΝ ΣΤΡΕΦΟΝΤΑΙ ΕΝΑΝΤΙΟΝ ΤΟΥ ΣΩΜΑΤΟΣ
Το ανοσοποιητικό μας σύστημα καταστρέφει διάφορους εισβολείς (μικροοργανισμούς, ουσίες κλπ.) που μπαίνουν στο σώμα μας και επιπλέον διατηρεί μνήμη για την περίπτωση επόμενης εισβολής του ιδίου μικροοργανισμού.
Όμως για να μην καταστρέφει και τις δικές μας πρωτεΐνες, όσα λεμφοκύτταρα στρέφονται εναντίον μας εξουδετερώνονται.
Η ικανότητα του ανοσοποιητικού συστήματος να διαχωρίζει μεταξύ των ξένων και των δικών μας πρωτεϊνών και να ΜΗ θεωρεί σαν ξένες και να ανέχεται τις δικές μας πρωτεΐνες ονομάζεται ανοσολογική ανοχή (Immune tolerance).
Τα Τ και Β λεμφοκύτταρα έχουν εκατομμύρια διαφορετικούς υποδοχείς (TCR τα Τ / BCR τα Β) που δημιουργούνται με τυχαίο τρόπο (με τυχαίο ανασυνδυασμό γονιδίων ή V(D)J recombination) στο θύμο αδένα τα Τ, στο μυελό των οστών τα Β.
Αυτό συμβαίνει για να μπορούν να αναγνωρίζουν όλες τις μελλοντικές απειλές από ξένες πρωτεΐνες εισβολέων στο σώμα μας. Όμως αναπόφευκτα, λόγω του τυχαίου τρόπου δημιουργίας τους, δυστυχώς πολλοί από τους υποδοχείς των Β και Τ λεμφοκυττάρων, στρέφονται και εναντίον δικών μας πρωτεϊνών.
Έτσι η φύση, ο Θεός, έπρεπε να εξουδετερώσουν αυτά τα Β και Τ λεμφοκύτταρα που οι υποδοχείς τους στρέφονται εναντίον μας.
Αυτό γίνεται σε πρώτη φάση στο θύμο αδένα για τα Τ και στο μυελό των οστών για τα Β λεμφοκύτταρα και λέγεται κεντρική ανοσολογική ανοχή (central tolerance), δηλαδή στην ουσία γίνεται ¨εξουδετέρωση¨ όσων λεμφοκυττάρων στρέφονται εναντίον μας.
Αργότερα στους λεμφαδένες και στον σπλήνα, γίνεται δεύτερη “εξουδετέρωση” (peripheral tolerance) όσων λεμφοκυττάρων στρέφονται εναντίον μας και ξέφυγαν από την πρώτη “εξουδετέρωση”. (Λεπτομέρειες υπάρχουν στο τέλος)
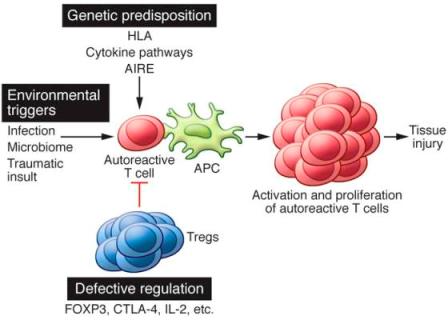
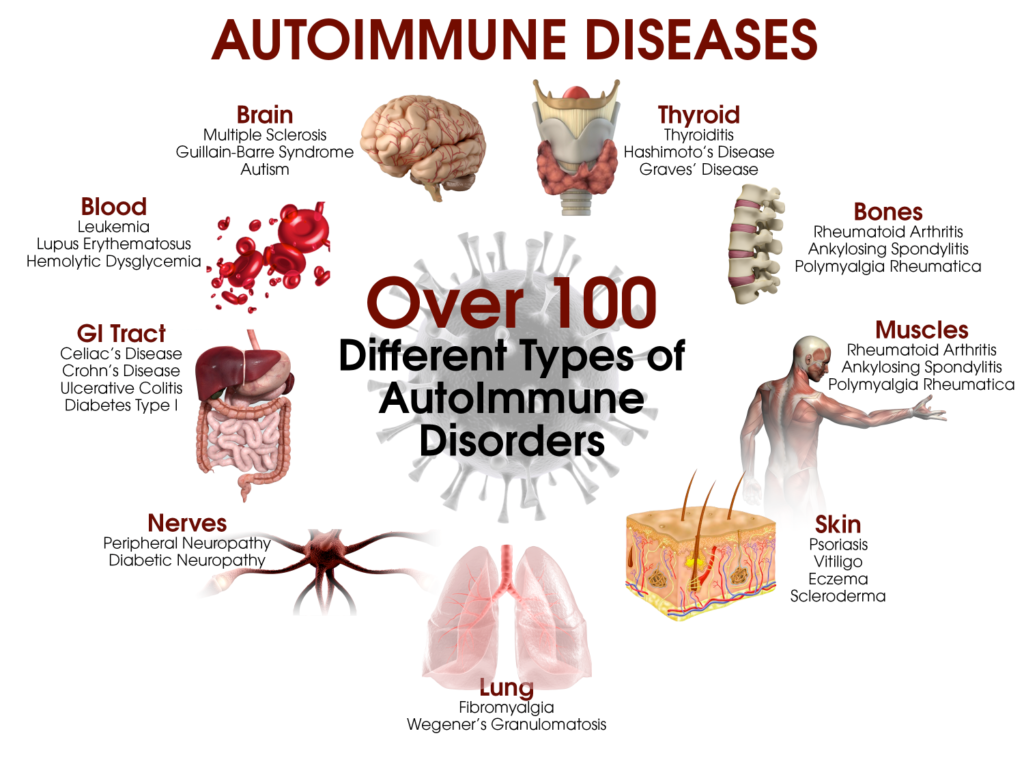
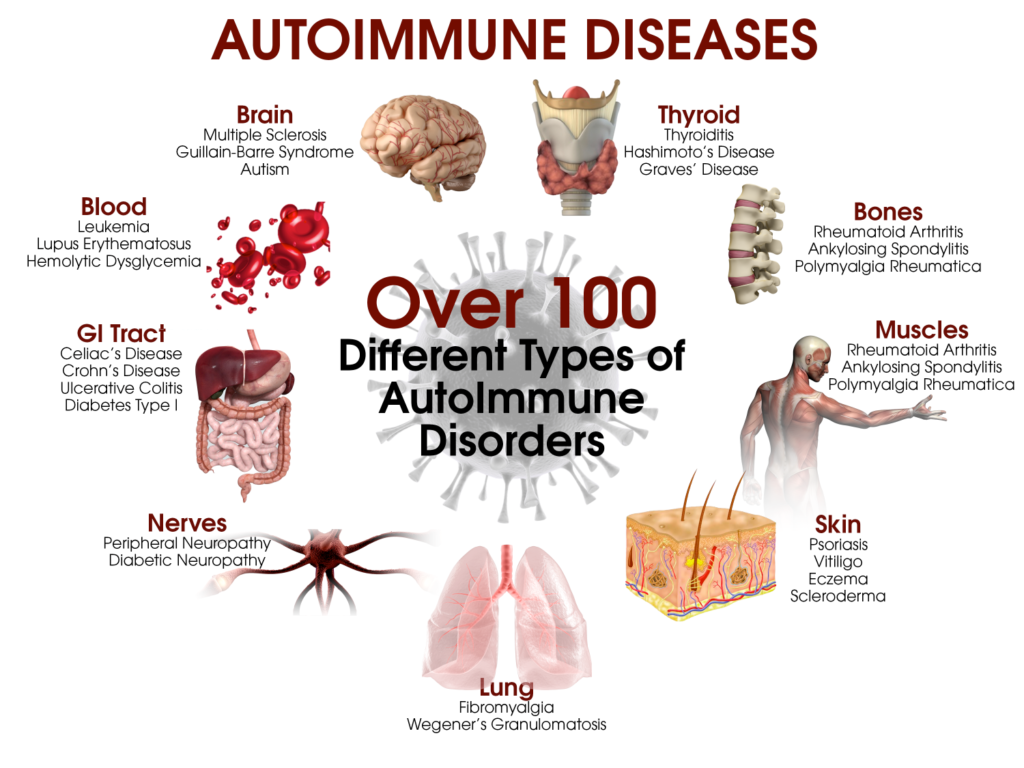
>> Ανάλογα με τα κύτταρα που προσβάλλονται δημιουργούνται περίπου 100 διαφορετικές αυτοάνοσες παθήσεις και η προσβολή μπορεί να αφορά ένα όργανο (π.χ. θυρεοειδίτιδα του Hashimoto, Διαβήτης τύπου 1, Λεύκη κλπ.) ή πολλά όργανα (π.χ. Ρευματοειδής αρθρίτιδα, συστηματικός Ερυθηματώδης λύκος κλπ.).
>> Σε πολλές από τις αυτοάνοσες παθήσεις προκαλείται και βλάβη στην καρδιά ή/και τα αγγεία. Η βλάβη μπορεί να είναι επιτάχυνση της αθηρωμάτωσης, καρδιακή ανεπάρκεια από βλάβη του μυ, περικαρδίτιδα, βλάβη βαλβίδων (π.χ. ανεπάρκεια αορτικής βαλβίδας), αρρυθμίες, διαταραχές αγωγής του ηλεκτρισμού, πνευμονική αρτηριακή υπέρταση κλπ.
ΟΙ ΑΥΤΟΑΝΟΣΕΣ ΠΑΘΗΣΕΙΣ
Περίπου το 6% του πληθυσμού και περισσότερο οι γυναίκες (5:1 συγκριτικά με τους άντρες), έχει κάποια αυτοάνοση πάθηση που συνήθως ξεκινά στην ενήλικη ζωή.
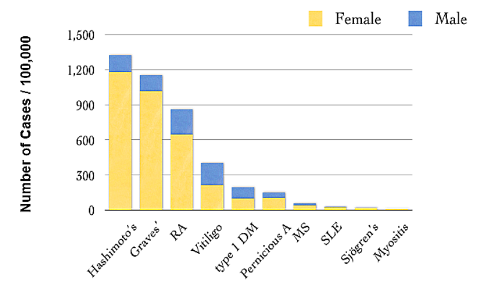
Μερικές από τις γνωστότερες αυτοάνοσες παθήσεις είναι ο υποθυρεοειδισμός από Θυρεοειδίτιδα του Hashimoto, ο υπερθυρεοειδισμός (από νόσο του Graves), ο Σ. Διαβήτης τύπου Ι, η Ρευματοειδής αρθρίτιδα, ο Ερυθηματώδης λύκος, η Αγκυλοποιητική σπονδυλαρθρίτιδα, η Σκληροδερμία (Συστηματική Σκλήρυνση) , η Δερματομυοσίτιδα, το σύνδρομο Sjögren, η νόσος του Addison κλπ.
Μερικές ακόμη αυτοάνοσες παθήσεις είναι, η κοιλιοκάκη, η ελκώδης κολίτιδα, η κακοήθης αναιμία, η απλαστική αναιμία, η λεύκη, η ψωρίαση, η μυασθένεια Gravis, η πολλαπλή σκλήρυνση (MS ή Σκλήρυνση κατά Πλάκας) που προσβάλλει το κεντρικό νευρικό σύστημα, η πέμφιγα, και άλλες περίπου 100 αυτοάνοσες παθήσεις.
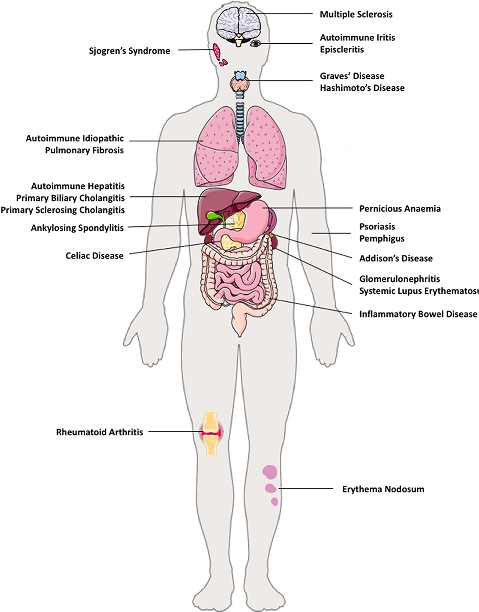
ΣΥΜΠΤΩΜΑΤΑ ΑΠΟ ΤΙΣ ΑΥΤΟΑΝΟΣΕΣ ΠΑΘΗΣΕΙΣ
Τα συμπτώματα ποικίλουν ανάλογα με την πάθηση, όμως μπορεί να υπάρχουν: Δεκατική πυρετική κίνηση, εξάντληση, πόνοι σε μυς ή/και σε αρθρώσεις, εξάνθημα κλπ. Τα συμπτώματα μπορεί να υποχωρούν και να ξαναεμφανίζονται κατά περιόδους.
ΛΙΓΑ ΓΙΑ ΟΡΙΣΜΕΝΕΣ ΑΥΤΟΑΝΟΣΕΣ ΠΑΘΗΣΕΙΣ
ΡΕΥΜΑΤΟΕΙΔΗΣ ΑΡΘΡΙΤΙΔΑ
Η Ρευματοειδής αρθρίτιδα είναι η συχνότερη χρόνια φλεγμονώδης πολυαρθρίτιδα και εμφανίζεται περίπου στο 1% των ενηλίκων, κυρίως στις γυναίκες (σε αναλογία 3:1 με τους άνδρες), κυρίως μεταξύ 20 και 50 χρονών.
Αυτή είναι συμμετρική πολυαρθρίτιδα μικρών και μεγάλων αρθρώσεων που παρουσιάζει καταστροφή των οστών της άρθρωσης και υπερπλασία της αρθρικής μεμβράνης.
Οι καρδιακές εκδηλώσεις της μπορεί να είναι περικαρδίτιδα (40%), μυοκαρδίτιδα, αυξημένη πιθανότητα στεφανιαίας νόσου (περίπου 50%), καρδιακής ανεπάρκειας, ανεπάρκειας μιτροειδούς βαλβίδας, κολπικών αρρυθμιών κλπ.
ΑΓΓΥΛΟΠΟΙΗΤΙΚΗ ΣΠΟΝΔΥΛΑΡΘΡΙΤΙΔΑ (ΑΓΚΥΛΩΤΙΚΗ ΣΠΟΝΔΥΛΙΤΙΔΑ)
Η Αγκυλοποιητική Σπονδυλαρθρίτιδα είναι χρόνια συστηματική φλεγμονή των συνδέσμων της σπονδυλικής στήλης, των ιερολαγονίων αρθρώσεων και άλλων περιφερικών αρθρώσεων (συνήθως δεν είναι αμφοτερόπλευρες), εμφανίζεται περίπου σε 1 ανά 200 ενήλικες (με ίδια συχνότητα σε άντρες και γυναίκες).
Οι καρδιακές επιπλοκές της αφορούν φλεγμονή στην αορτή, ανεπάρκεια αορτικής (και μιτροειδούς) βαλβίδας, διαταραχές στη μετάδοση του ηλεκτρισμού κυρίως στον κολποκοιλιακό κόμβο, βλάβη στο μυ της καρδιάς κλπ.
ΣΥΣΤΗΜΑΤΙΚΟΣ ΕΡΥΘΗΜΑΤΩΔΗΣ ΛΥΚΟΣ
Ο Συστηματικός Ερυθηματώδης Λύκος είναι χρόνια φλεγμονώδης νόσος πολλών συστημάτων του σώματος που εμφανίζεται περίπου σε 1 ανά 2.000 ενήλικες, κυρίως στις γυναίκες (σε αναλογία 9:1 με τους άνδρες). Μπορεί να προκαλέσει βλάβη σε πολλά όργανα, όπως στις αρθρώσεις, στα νεφρά, στο νευρικό σύστημα (κεντρικό και περιφερικό), εξάνθημα κλπ.
Επίσης μπορεί να προκαλέσει δευτεροπαθές αντιφωσφολιπιδικό σύνδρομο με θρομβώσεις, αυτόματες αποβολές κλπ.
Οι καρδιακές εκδηλώσεις του μπορεί να είναι περικαρδίτιδα (50%), ενδοκαρδίτιδα Libman-Sacks και βαλβιδικές ανωμαλίες (50%), μυοκαρδίτιδα, διπλάσια πιθανότητα στεφανιαίας νόσου, αυξημένη πιθανότητα καρδιακής ανεπάρκειας, κολπικών αρρυθμιών, π.χ. φλεβοκομβική ταχυκαρδία, διαταραχές αγωγής, συχνά ήπια πνευμονική αρτηριακή υπέρταση κλπ.
ΣΚΛΗΡΟΔΕΡΜΙΑ (ΣΥΣΤΗΜΑΤΙΚΗ ΣΚΛΗΡΥΝΣΗ)
Το διάχυτο Σκληρόδερμα είναι σπάνια αυτοάνοση πάθηση που προκαλεί εναπόθεση κολλαγόνου στο δέρμα και τα εσωτερικά όργανα (καρδιά, νεφρά, πνεύμονες, γαστρεντερικό κλπ.) και επιπλέον βλάβη των μικρών αρτηριών.
Αυτή εμφανίζεται περίπου σε 1 ανά 5.000 ενήλικες (νέα εμφάνιση 150 περίπου άνθρωποι ανά έτος στην Ελλάδα), κυρίως σε γυναίκες (σε αναλογία περίπου 4 προς 1 με τους άνδρες). Η έναρξη της συνήθως γίνεται σε ηλικία 30 ως 65 ετών.
Από πλευράς καρδιάς συνήθως υπάρχει υγρό στο περικάρδιο, ίνωση στην καρδιά με επακόλουθο καρδιακή ανεπάρκεια, διαταραχές αγωγής ηλεκτρισμού, έκτακτες κοιλιακές συστολές, βλάβη των αρτηριολίων στην καρδιά με συνέπεια καρδιακή ανεπάρκεια και στα πνευμονικά αρτηριόλια με συνέπεια πνευμονική αρτηριακή υπέρταση.
Επιπλέον συνήθως συνυπάρχει το φαινόμενο Raynaud’s στα χέρια (και πόδια) από σπασμό των αρτηριολίων λόγω κρύου και από άγχος.
Η ΚΟΚΚΙΩΜΑΤΩΣΗ ΜΕ ΠΟΛΥΑΓΓΕΙΙΤΙΔΑ (WEGENER)
Η κοκκιωμάτωση με πολυαγγειίτιδα (GPA) είναι μια σπάνια αυτοάνοση πάθηση όπου το ανοσοποιητικό επιτίθεται και προκαλεί βλάβη στο ενδοθήλιο των μικρών αγγείων σε όλο το σώμα. (Αυτή λεγόταν αγγειίτιδα Wegener)
Η κοκκιωμάτωση με πολυαγγειίτιδα παρατηρείται περίπου σε 1 ανά 5.000 ανθρώπους και εμφανίζεται ιδίως σε ηλικίες 35 ως 65 ετών, με ίδια συχνότητα σε άντρες και γυναίκες.
Το αποτέλεσμα είναι ότι μειώνεται η αιμάτωση σχεδόν σε όλα τα όργανα (και εμφανίζονται νεκρωτικές κοκκιωματώδεις μάζες) με συχνότερη εντόπιση τους πνεύμονες, το ανώτερο αναπνευστικό και τα νεφρά. (Επιπλέον μπορεί να εμφανιστεί θρομβοεμβολική νόσος)
Τα συμπτώματα εξαρτώνται από τα όργανα που έχουν προσβληθεί και είναι συνηθέστερα βήχας, δύσπνοια, ρινική συμφόρηση, πυρετική κίνηση, εξάντληση και απώλεια βάρους.
Τα αυτοαντισώματα που ανιχνεύονται είναι συνηθέστερα τα ANCA (anti neutrophil cytoplasmic antibodies ή αντιουδετεροφιλικά κυτοπλασματικά αντισώματα), όμως συνήθως χρειάζεται βιοψία σε κοκκιωματώδεις μάζες για να μπει η διάγνωση της.
Η σύγχρονη θεραπεία για την ελαφρά νόσο είναι η Μethotrexate + Κορτιζόνη και για την βαρειά νόσο είναι η Rituximab* + Κορτιζόνη. Επίσης για τη βαρειά νόσο έχει εγκριθεί το Avacopan – Tanveos (αποκλειστής του υποδοχέα του συμπληρώματος C5a).
Το Rituximab* (MabThera) είναι μονοκλωνικό αντίσωμα κατά των Β λεμφοκυττάρων (anti-CD20 antibody)
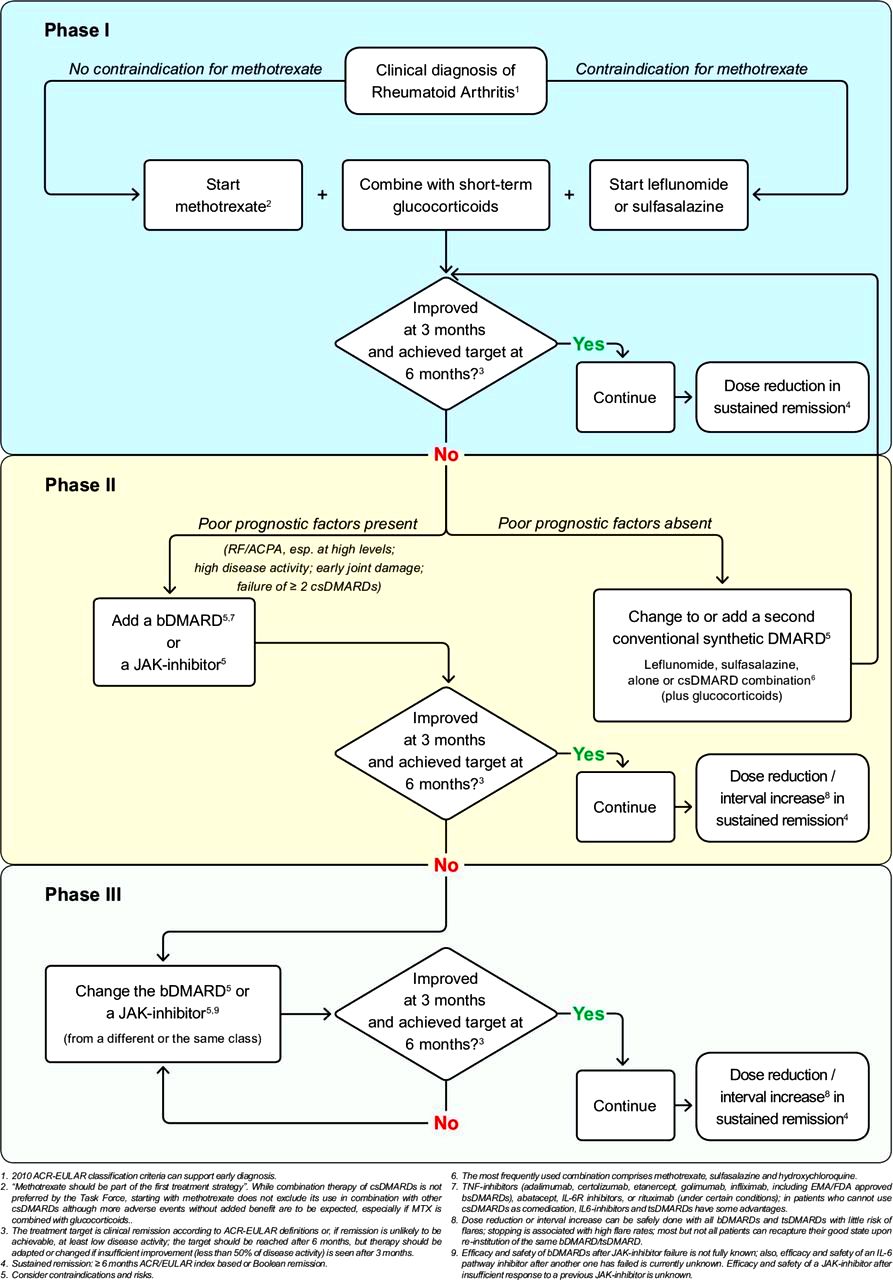
Η ΘΕΡΑΠΕΙΑ ΤΗΣ ΡΕΥΜΑΤΟΕΙΔΟΥΣ ΑΡΘΡΙΤΙΔΑΣ
Η θεραπεία, πέρα από τις υγιεινο-διαιτητικές αλλαγές, γίνεται με αντιρευματικά φάρμακα που τροποποιούν τη νόσο (disease-modifying antirheumatic drugs ή DMARDs) και εξαρτάται κυρίως από τη βαρύτητα της πάθησης και από διάφορους παράγοντες του ασθενούς.
[Η βιταμίνη D (2000 IU/μέρα) σε ανθρώπους μεγαλύτερους των 50-55 ετών, ίσως μειώνει τη μελλοντική εμφάνιση αρκετών αυτοάνοσων παθήσεων κατά 22%, μετά από 5 χρόνια χορήγησης. (Δρα σε κύτταρα της εγγενούς και επίκτητης ανοσίας, με τελικό αποτέλεσμα τη μείωση φλεγμονωδών κυτταροκινών και αυτοαντισωμάτων)
https://www.bmj.com/content/376/bmj-2021-066452]
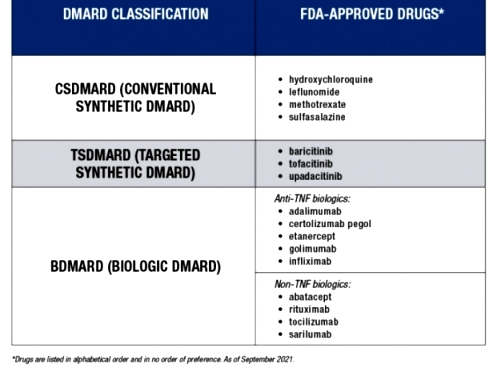
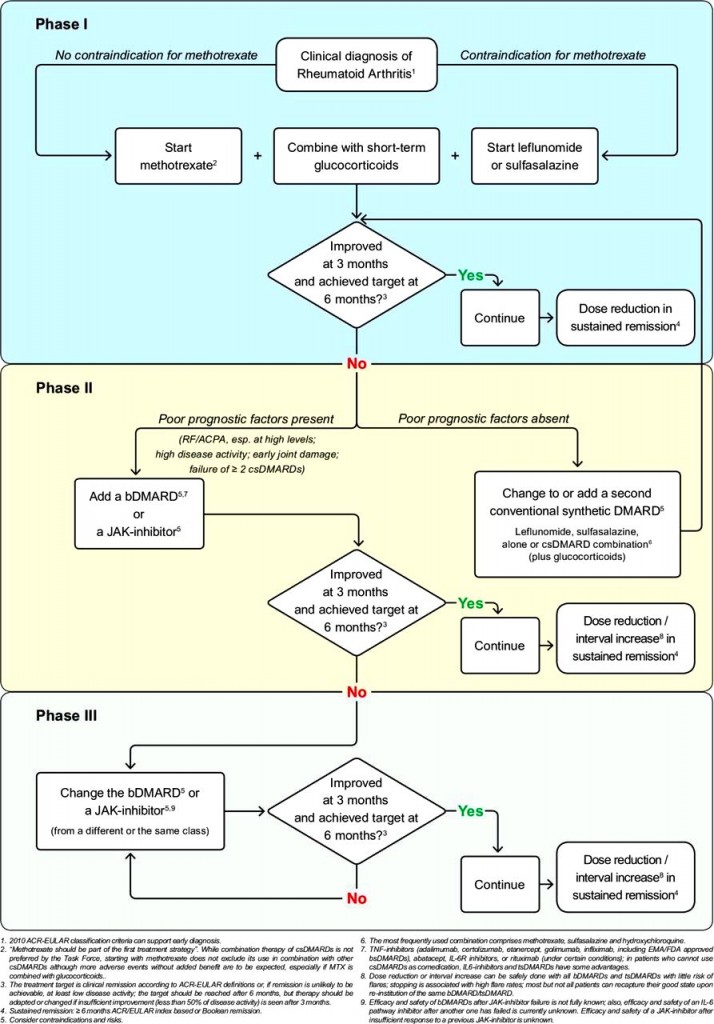
>>>> Η αρχική θεραπεία ξεκινά με φάρμακα που μειώνουν τη δράση του ανοσοποιητικού συνολικά (csDMARDs): Αρχίζουμε με Μethotrexate και αν αυτή αντενδείκνυται μπορεί να χορηγηθεί Leflunomide ή Sulfasalazine (ή Hydroxychloroquine σε ελαφρά νόσο ή σε επάνοδο της), με ή χωρίς Κορτιζόνη (για λίγο, μέχρι να δράσουν τα υπόλοιπα).
[Αν δεν είναι ικανοποιητικό το αποτέλεσμα ίσως μπορεί να χορηγηθεί συνδυασμός methotrexate + sulfasalazine + hydroxychloroquine]
>>> Αν σε 3-6 μήνες δεν υπάρχει αποτέλεσμα με 2 τουλάχιστον από τα csDMARDs, μαζί με τη Methotrexate προσθέτουμε tsDMARDs όπως αναστολείς Κινάσης (JAK inhibitors)* ή προσθέτουμε βιολογικά bDMARDs, και ιδίως τους αναστολείς TNF** (π.χ. η Tofacitinib αυξάνει τους καρκίνους και τα καρδιαγγειακά επεισόδια συγκριτικά με τους αναστολείς TNF).
[*Οι tsDMARDs JAK inhibitors είναι η Upadacitinib (Rinvoq), η Baricitinib (Olumiant), η Tofacitinib (Xeljanz), η Filgotinib (Jyseleca).
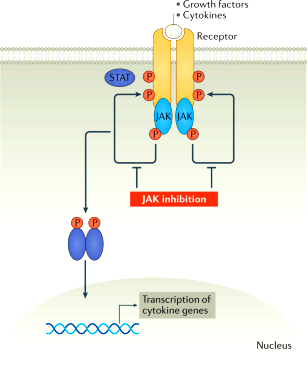
**Οι TNF inhibitors είναι μονοκλωνικά αντισώματα π.χ. η Infliximab (Remicade), η Adalimumab (Humira), η Certolizumab (Cimzia), η Golimumab (Simponi) ή η πρωτεΐνη (circulating receptor fusion protein) Etanercept (Enbrel)]
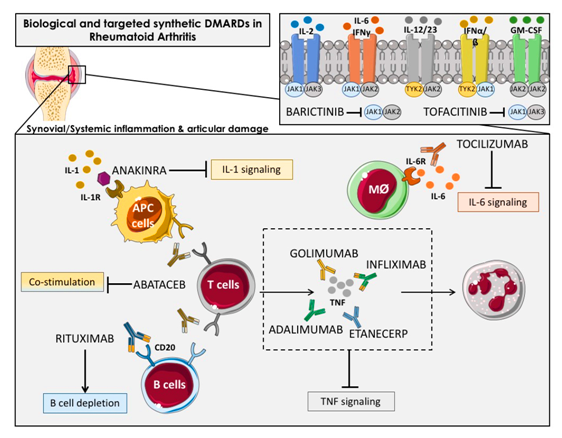
>> Σε βαρειά νόσο (αν δεν υπάρχει ικανοποιητικό αποτέλεσμα από τα προηγούμενα), μπορεί επίσης να χορηγηθεί Rituximab (MabThera) (μονοκλωνικό αντίσωμα κατά των Β λεμφοκυττάρων, anti-CD20 antibody) μαζί με Methotrexate.
Σε βαρειά νόσο (αν δεν υπάρχει ικανοποιητικό αποτέλεσμα από τα προηγούμενα) μπορεί να χορηγηθεί αναστολέας της Ιντερλευκίνης 6 όπως η Sarilumab (Kevzara) (ή η Tocilizumab / RoActemra) μαζί με Methotrexate.
Σε βαρειά νόσο μπορεί επίσης να χορηγηθεί Abatacept (Orencia) (αποκλείει τη δράση των Τ λεμφοκυττάρων, T cell costimulatory inhibitor) μαζί με Methotrexate.
Το Anakinra (Κineret) (αποκλειστής της Ιντελευκίνης 1) μπορεί να χορηγηθεί στα πλαίσια ιατρικής μελέτης.
ΟΙ ΑΙΤΙΕΣ ΤΩΝ ΑΥΤΟΑΝΟΣΩΝ ΠΑΘΗΣΕΩΝ ΛΕΠΤΟΜΕΡΕΣΤΕΡΑ
Οι αυτοάνοσες παθήσεις ξεκινούν να δημιουργούνται λόγω του ότι συνυπάρχουν ταυτόχρονα η πρωτεΐνη στόχος (λέγεται αυτοαντιγόνο) και μια προδιάθεση του ανοσοποιητικού συστήματος να στραφεί εναντίον των δικών του πρωτεϊνών.
>> Οι αυτοάνοσες παθήσεις οφείλονται σε γονιδιακές παραλλαγές – μεταλλάξεις ή/και σε επιγενετικές (περιβαλλοντικές) αιτίες.
> Οι γονιδιακές μεταλλάξεις μπορεί να συμβούν στα Τ και Β λεμφοκύτταρα μας ή σε γονίδιο αντιγόνου ιστοσυμβατότητας** (HLA ή MHC) ή σε γονίδιο της πρωτεΐνης autoimmune regulator*** ή σε γονίδιο κυτταροκίνης (ή υποδοχέα κυτταροκίνης) ή σε γονίδιο που κάνει μια παραγόμενη πρωτεΐνη να μοιάζει με ξένη]
> Οι επιγενετικές αιτίες μπορεί να είναι μια λοίμωξη από ιό ή μικρόβιο, ορισμένα χημικά στη διατροφή ή στον εισπνεόμενο αέρα, ορισμένα φάρμακα, αλλαγές στο μικροβίωμα του εντέρου (λόγω της σύγχρονης υγιεινής διαβίωσης), ένα οξειδωτικού stress κλπ.
>>> 1) Από πλευράς του ανοσοποιητικού συστήματος οι αυτοάνοσες παθήσεις δημιουργούνται λόγω του ότι:
α) Σταματά η κεντρική και η περιφερική αναγνώριση – ανοχή (tolerance) και εξουδετέρωση των λεμφοκυτάρων Τ και Β που οι υποδοχείς τους στρέφονται εναντίον των δικών μας πρωτεϊνών.
[Τα Τ και Β λεμφοκύτταρα έχουν εκατομμύρια διαφορετικούς υποδοχείς (TCR τα Τ, BCR τα Β) που δημιουργούνται με τυχαίο τρόπο (με τυχαίο ανασυνδυασμό γονιδίων ή V(D)J recombination) στο θύμο αδένα τα Τ, στο μυελό των οστών τα Β.
Αυτό συμβαίνει για να μπορούν να αναγνωρίζουν όλες τις μελλοντικές απειλές από ξένες πρωτεΐνες εισβολέων στο σώμα μας. Όμως αναπόφευκτα, λόγω του τυχαίου τρόπου δημιουργίας τους, δυστυχώς μερικοί από τους υποδοχείς των Β ή Τ λεμφοκυττάρων, στρέφονται και εναντίον δικών μας πρωτεϊνών.
Έτσι η φύση, ο Θεός, έπρεπε να εξουδετερώσουν αυτά τα Β και Τ λεμφοκύτταρα που οι υποδοχείς τους στρέφονται και εναντίον δικών μας πρωτεϊνών.
Η ικανότητα του ανοσοποιητικού συστήματος να ΜΗ θεωρεί σαν ξένες και να μην αντιδρά, να ανέχεται τις δικές μας πρωτεΐνες ονομάζεται ανοσολογική ανοχή (immunological tolerance).
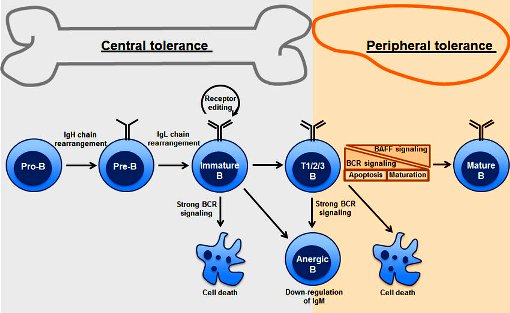
Αυτό γίνεται αρχικά στο στο θύμο αδένα για τα Τ και στο μυελό των οστών για τα Β λεμφοκύτταρα και λέγεται κεντρική ανοσολογική ανοχή (central tolerance) δηλαδή αναγνώριση και εξουδετέρωση των λεμφοκυττάρων που οι υποδοχείς τους στρέφονται και εναντίον των δικών μας πρωτεϊνών.
Αργότερα γίνεται στους λεμφαδένες και στο σπλήνα για όσα λεμφοκύτταρα ξεφύγουν και λέγεται περιφερική ανοσολογική ανοχή (peripheral tolerance), δηλαδή περιφερική αναγνώριση και εξουδετέρωση των λεμφοκυττάρων που οι υποδοχείς τους στρέφονται και εναντίον των δικών μας πρωτεϊνών.
Υπ’ όψιν ότι υπάρχουν πολλά σημεία ελέγχου της ανοσολογικής ανοχής (self-tolerance checkpoints) τόσο κεντρικά όσο και περιφερικά.]
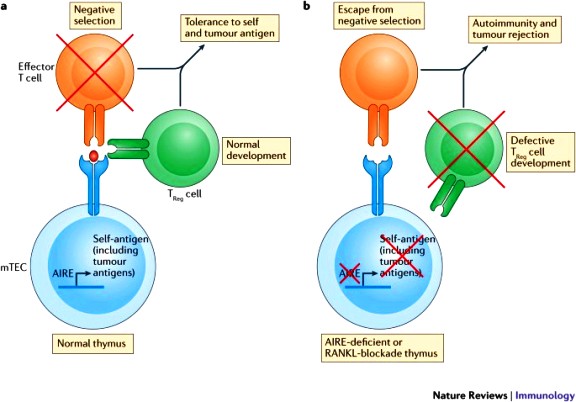
i) Η κεντρική αναγνώριση και εξουδετέρωση (central tolerance) συμβαίνει στο θύμο αδένα για τα Τ λεμφοκύτταρα [με τη βοήθεια και της πρωτεΐνης autoimmune regulator*** (AIRE), και του άξονα RANK/RANKL] και στο μυελό των οστών για τα Β λεμφοκύτταρα.
[Ο άξονας RANK/RANKL δρα μέσω των μυελωδών επιθηλιακών κυττάρων του θύμου αδένα (mTECs)]
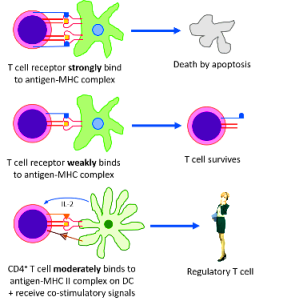
>> Έτσι όσα ανώριμα Τ λεμφοκύτταρα βρεθούν ότι ενώνονται ΙΣΧΥΡΑ στις πρωτεΐνες του σώματος μας που εμφανίζονται στις πρωτεΐνες MHC Ι και MHC ΙΙ** στην επιφάνεια των επιθηλιακών (mTECs), των δενδριτικών κυττάρων και των Β λεμφοκυττάρων του θύμου, καταστρέφονται ώστε να μην μπουν στην κυκλοφορία (negative selection).
>> Με παρόμοιο μηχανισμό καταστρέφονται και όσα ανώριμα Β λεμφοκύτταρα βρεθούν ότι ενώνονται ΙΣΧΥΡΑ στις πρωτεΐνες του σώματος μας που εμφανίζονται στα MHC Ι και MHC ΙΙ** δενδριτικών κυττάρων στο μυελό των οστών.
Αν βρεθεί ότι ο υποδοχέας των Β λεμφοκυττάρων, BCR, συνδέεται με πρωτεΐνες του σώματος μας (αυτοαντιγόνα) στο μυελό των οστών, θα υποστεί θάνατο ή θα γίνει αλλαγή του υποδοχέα με κόψιμο, αναδιάταξη και ράψιμο των γονιδίων του (V(D)J recombination).
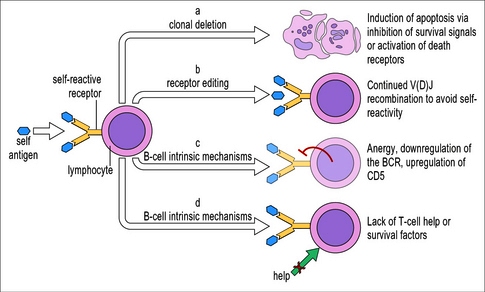
ii) Η περιφερική αναγνώριση και εξουδετέρωση (peripheral tolerance) γίνεται στους λεμφαδένες, τον σπλήνα και τοπικά σε άλλους ιστούς, για όσα λεμφοκύτταρα ξέφυγαν από την κεντρική εξουδετέρωση.
Αυτή γίνεται είτε με άμεση απενεργοποίηση ή θάνατο (clonal deletion) τους είτε από τα ρυθμιστικά – ανοσοκατασταλτικά λεμφοκύτταρα Tregs και Bregs (επίσης από ανασταλτικές Κυτταροκίνες, από κύτταρα της εγγενούς ανοσίας κλπ.)
Επιπλέον όσα Β λεμφοκύτταρα δεν δεχθούν διεγερτικά σήματα από TFH λεμφοκύτταρα υφίστανται θάνατο. (Όσα από τα TFH λεμφοκύτταρα ενώνονταν ισχυρά με δικά μας αντιγόνα εξαλείφθηκαν κεντρικά στο θύμο αδένα).
[Τα Tregs (Regulatory T) είναι υποπληθυσμός των Τ λεμφοκυττάρων που δημιουργείται στο θύμο αδένα και τα Bregs (Regulatory B), δημιουργούνται στον μυελό των οστών και εκκρίνουν μεταξύ άλλων ανοσοκατασταλτικών ουσιών, και Ιντερλευκίνη 10 (B10 Bregs).
Τα Tregs και τα Bregs καταστέλλουν το ανοσοποιητικό σύστημα όταν έχει εξοντωθεί ο εισβολέας και επιπλέον βοηθούν στην πρόληψη αυτοάνοσων παθήσεων.
Η λειτουργία των Tregs είναι κυρίως να μειώνουν τον πολλαπλασιασμό, την ενεργοποίηση και την παραγωγή κυτταροκινών των CD4+ T και CD8+ T λεμφοκυττάρων.
Αυτό γίνεται από τον πυρηνικό παράγοντα αντιγραφής FoxP3 που περιέχουν, κυρίως μέσω της παραγωγής Ιντερλευκίνης 10 (IL 10) και του παράγοντα TGF-β.
>> Όταν υπολειτουργούν/δυσλειτουργούν τα Tregs και τα Bregs, δεν μειώνονται τα Τ λεμφοκύτταρα (CD4+ και CD8+) και τα Β λεμφοκύτταρα αντίστοιχα, που στρέφονται εναντίον του σώματος, οπότε εμφανίζονται αυτοάνοσες παθήσεις.
(Αντίθετα όταν αυτά υπερλειτουργούν ευνοείται η δημιουργία καρκίνου, λόγω μείωσης της κατασταλτικής δράσης του ανοσοποιητικού απέναντι στα καρκινικά κύτταρα.)]
β) Λόγω μεταλλάξεων σε Τ και Β λεμφοκύτταρα μας, κατά τη διαδικασία της κλωνοποίησης τους, που αυξάνονται με την πάροδο της ηλικίας.
>>> 2) Όμως μπορεί και οι δικές μας πρωτεΐνες να τροποποιηθούν:
α) είτε λόγω μετάλλαξης σε γονίδιο που κάνει την παραγόμενη πρωτεΐνη να μοιάζει με ξένη.
β) είτε λόγω φλεγμονής, ή οξειδωτικού stress ή λόγω λοίμωξης από μικρόβιο**** ή από ιό***** (π.χ. από τον ιό Coxsackie), οπότε τα κύτταρα μας αρχίζουν να εκκρίνουν φλεγμονώδεις ουσίες (κυτταροκίνες, Ιντερφερόνες) που προσελκύουν το ανοσοποιητικό μας σύστημα να τα εκλάβει σαν ξένα και να επιτεθεί εναντίον τους.
Επίσης σε λοίμωξη ή τραύμα π.χ. στον οφθαλμό, απελευθερώνονται κρυμμένες πρωτεΐνες οπότε το ανοσοποιητικό στρέφεται εναντίον τους.
[**Τα αντιγόνα ιστοσυμβατότητας (Human Leucocyte Antigen ή HLA) λέγονται και σύμπλεγμα μείζονος ιστοσυμβατότητας (major histocombatibility complex ή MHC), είναι γλυκοπρωτεΐνες στην επιφάνεια των κυττάρων μας και είναι τα “σημάδια” ότι τα κύτταρα είναι δικά μας.
Οι πρωτεΐνες MHC είναι απαραίτητες για να διαχωρίζει το αμυντικό μας σύστημα, ποια μόρια – αντιγόνα είναι δικά μας (αυτοαντιγόνα) και πια είναι ξένα ή αν τα κύτταρα είναι φυσιολογικά ή μολυσμένα π.χ. από ιό, βακτήριο.
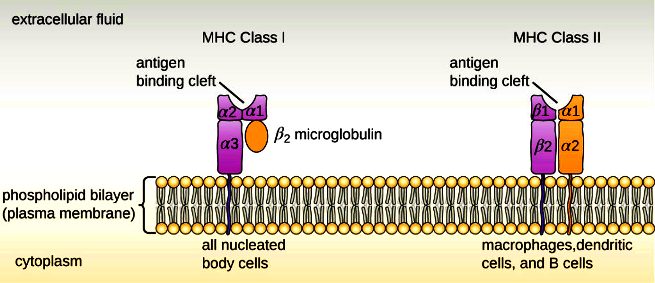
Επίσης σε περίπτωση μεταμόσχευσης οργάνου το αμυντικό σύστημα απορρίπτει το μόσχευμα, αν αυτό δεν έχει ίδια MHC ή HLA με του λήπτη
***Η πρωτεΐνη autoimmune regulator παρουσιάζει τις φυσιολογικές πρωτεΐνες του σώματος μας στα επιθηλιακά κύτταρα του θύμου αδένα (mTEC), έτσι ώστε όσα Τ λεμφοκύτταρα συνδεθούν ισχυρά με αυτές καταστρέφονται
****Παράδειγμα λοίμωξης από μικρόβιο είναι η λοίμωξη από Στρεπτόκοκκο στην παιδική ηλικία που προκαλεί προοδευτικά επιδεινούμενη Ρευματική βλάβη σε καρδιακές βαλβίδες.
Το ανοσοποιητικό σύστημα που στρέφεται κατά πρωτεϊνών του Στρεπτόκοκκου, στρέφεται και κατά της Μυοσίνης των καρδιακών βαλβίδων που μοιάζει με τις πρωτεΐνες του Στρεπτόκοκκου (molecular mimicry, cross-reactivity)
*****Παράδειγμα λοίμωξης από τον ιό Coxsackie Β είναι η πιθανή πρόκληση Σ. Διαβήτη τύπου 1.]
ΕΝΔΕΙΚΤΙΚΗ ΒΙΒΛΙΟΓΡΑΦΙΑ
https://www.sciencedirect.com/science/article/abs/pii/S000293431500443X
https://www.sciencedirect.com/science/article/abs/pii/S1568997221002007
https://ard.bmj.com/content/79/6/685
https://www.nature.com/articles/s41375-021-01231-3
https://www.pnas.org/content/116/18/9014
https://www.frontiersin.org/articles/10.3389/fimmu.2021.592914/full
https://www.frontiersin.org/articles/10.3389/fimmu.2021.731947/full
https://www.mdpi.com/1422-0067/21/23/9067/htm#
https://www.nature.com/articles/d42859-021-00026-x
https://www.nature.com/articles/nri.2017.19
https://www.nature.com/articles/nri.2016.9
https://www.revespcardiol.org/en-cardiovascular-disorders-rheumatic-disease-articulo-S1885585711004014
https://onlinelibrary.wiley.com/doi/full/10.1111/imm.12831
https://www.rheumatology.org/Portals/0/Files/2021-ACR-Guideline-for-Treatment-Rheumatoid-Arthritis-Early-View.pdf
https://www.frontiersin.org/articles/10.3389/fimmu.2020.623265/full
https://www.nature.com/articles/nri.2017.19
https://www.frontiersin.org/articles/10.3389/fimmu.2021.766698/full
https://www.mdpi.com/2073-4409/10/5/1190/htm
Drug treatment for rheumatoid arthritis – NICE Pathways



{kind=link}
{kind=link}